
Drug discovery has come a long way from its roots in serendipity. It is now an increasingly rational process, in which one important phase, called lead optimization, is the stepwise search for promising drug candidate compounds in the lab. In this phase, expert medicinal chemists work to improve “hit” molecules—compounds that demonstrate some promising properties, as well as some undesirable ones, in early screening. In subsequent testing, chemists try to adapt the structure of hit molecules to improve their biological efficacy and reduce potential side effects. This process combines knowledge, creativity, experience, and intuition, and often lasts for years. Over many decades, computational modelling techniques have been developed to help predict how the molecules will fare in the lab, so that costly and time-consuming experiments can focus on the most promising compounds.
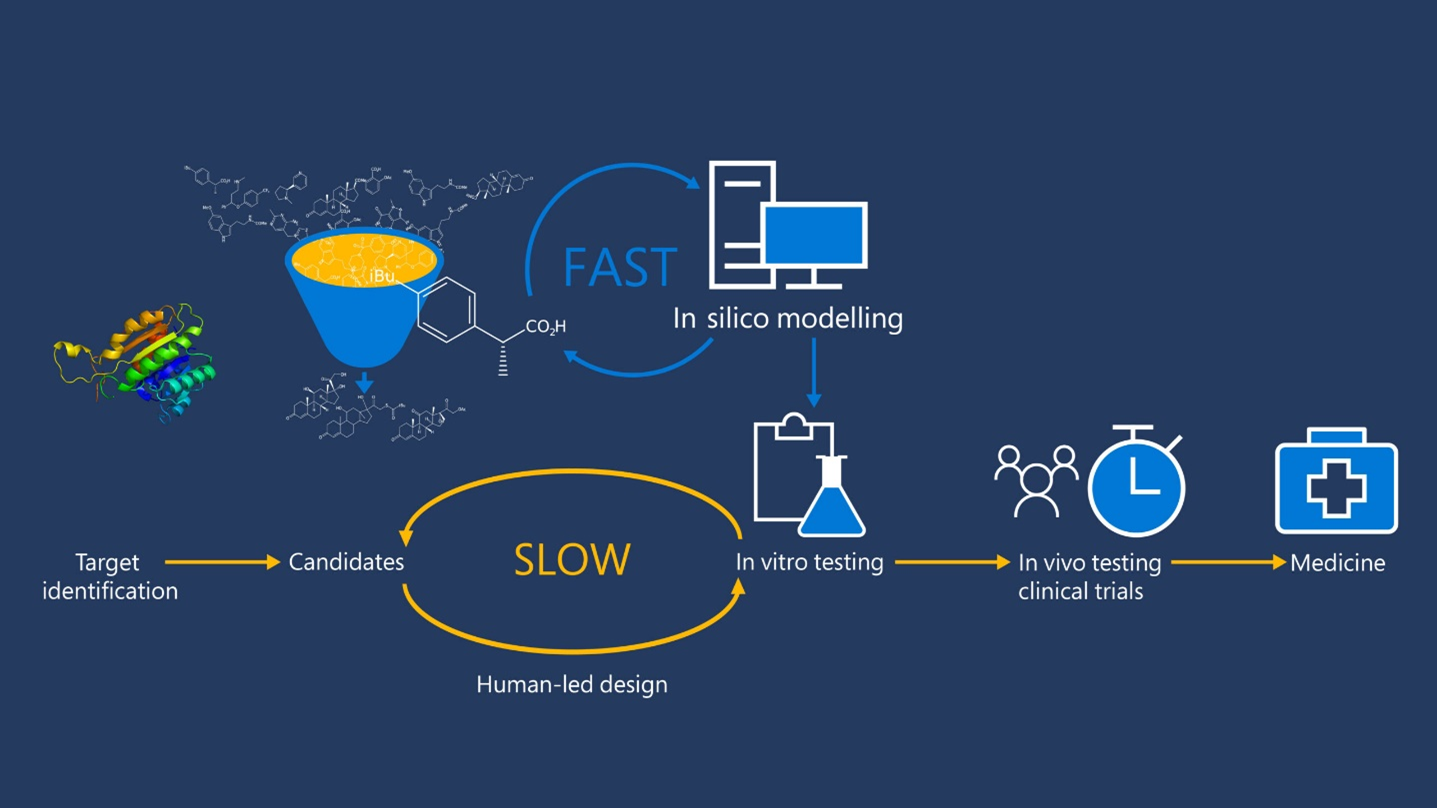
The Microsoft Generative Chemistry team is working with Novartis to improve these modelling techniques with a new model called MoLeR.
“MoLeR illustrates how generative models based on deep learning can help transform the drug discovery process and enable our colleagues at Novartis to increase the efficiency in finding new compounds.”
Christopher Bishop, Technical Fellow and Laboratory Director, Microsoft Research Cambridge
We recently focused on predicting molecular properties using machine learning methods in the FS-Mol project. To further support the drug discovery process, we are also working on methods that can automatically design compounds that better fit project requirements than existing candidate compounds. This is an extremely difficult task, as only a few promising molecules exist in the vast and largely unexplored chemical space—estimated to contain up to 1060 drug-like molecules. Just how big is that number? It would be enough molecules to reproduce the Earth billions of times. Finding them requires creativity and intuition that cannot be captured by fixed rules or hand-designed algorithms. This is why learning is crucial not only for the predictive task, as done in FS-Mol, but also for the generative task of coming up with new structures.
In our earlier work, published at the 2018 Conference on Neural Information Processing Systems (NeurIPS), we described a generative model of molecules called CGVAE. While that model performed well on simple, synthetic tasks, we noted then that further improvements required the expertise of drug discovery specialists. In collaboration with experts at Novartis, we identified two issues limiting the applicability of the CGVAE model in real drug discovery projects: it cannot be naturally constrained to explore only molecules containing a particular substructure (called the scaffold), and it struggles to reproduce key structures, such as complex ring systems, due to its low-level, atom-by-atom generative procedure. To remove these limitations, we built MoLeR, which we describe in our new paper, “Learning to Extend Molecular Scaffolds with Structural Motifs,” published at the 2022 International Conference on Learning Representations (ICLR).
The MoLeR model
In the MoLeR model, we represent molecules as graphs, in which atoms appear as vertices that are connected by edges corresponding to the bonds. Our model is trained in the auto-encoder paradigm, meaning that it consists of an encoder—a graph neural network (GNN) that aims to compress an input molecule into a so-called latent code—and a decoder, which tries to reconstruct the original molecule from this code. As the decoder needs to decompress a short encoding into a graph of arbitrary size, we design the reconstruction process to be sequential. In each step, we extend a partially generated graph by adding new atoms or bonds. A crucial feature of our model is that the decoder makes predictions at each step solely based on a partial graph and a latent code, rather than in dependence on earlier predictions. We also train MoLeR to construct the same molecule in a variety of different orders, as the construction order is an arbitrary choice.

As we alluded to earlier, drug molecules are not random combinations of atoms. They tend to be composed of larger structural motifs, much like sentences in a natural language are compositions of words, and not random sequences of letters. Thus, unlike CGVAE, MoLeR first discovers these common building blocks from data, and is then trained to extend a partial molecule using entire motifs (rather than single atoms). Consequently, MoLeR not only needs fewer steps to construct drug-like molecules, but its generation procedure also occurs in steps that are more akin to the way chemists think about the construction of molecules.
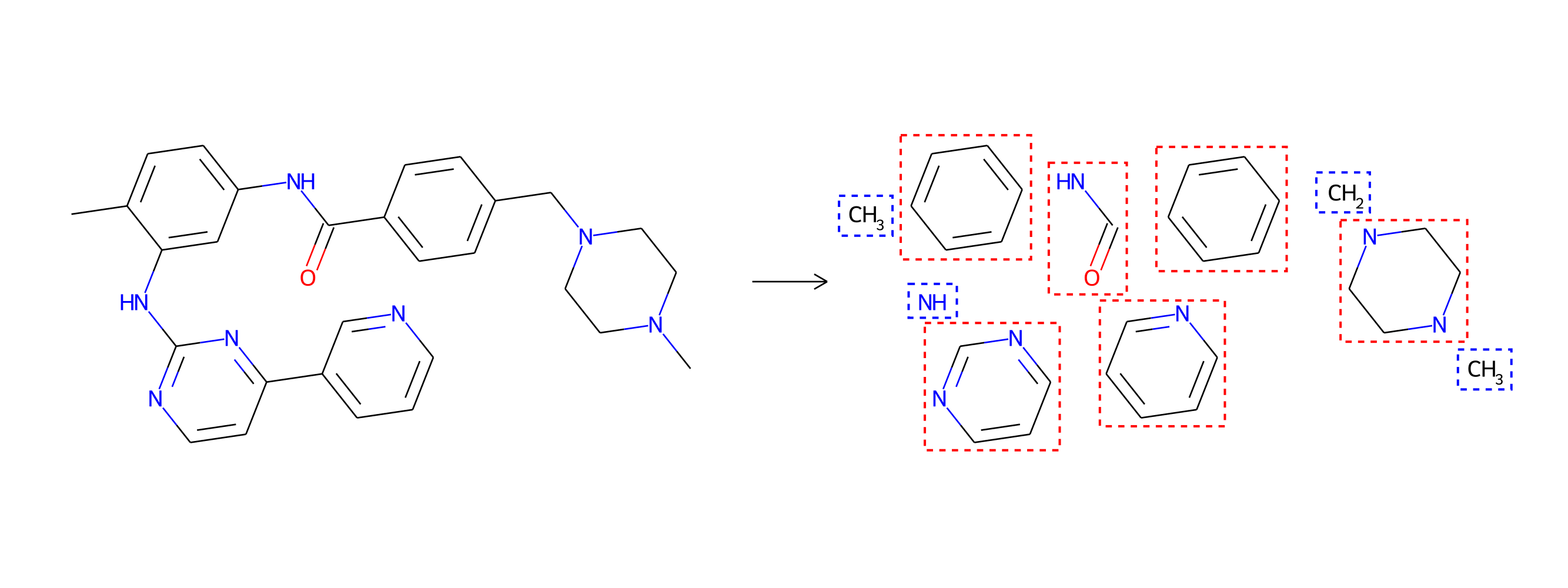
Drug-discovery projects often focus on a specific subset of the chemical space, by first defining a scaffold—a central part of the molecule that has already shown promising properties—and then exploring only those compounds that contain the scaffold as a subgraph. The design of MoLeR’s decoder allows us to seamlessly integrate an arbitrary scaffold by using it as an initial state in the decoding loop. As we randomize the generation order during training, MoLeR implicitly learns to complete arbitrary subgraphs, making it ideal for focused scaffold-based exploration.
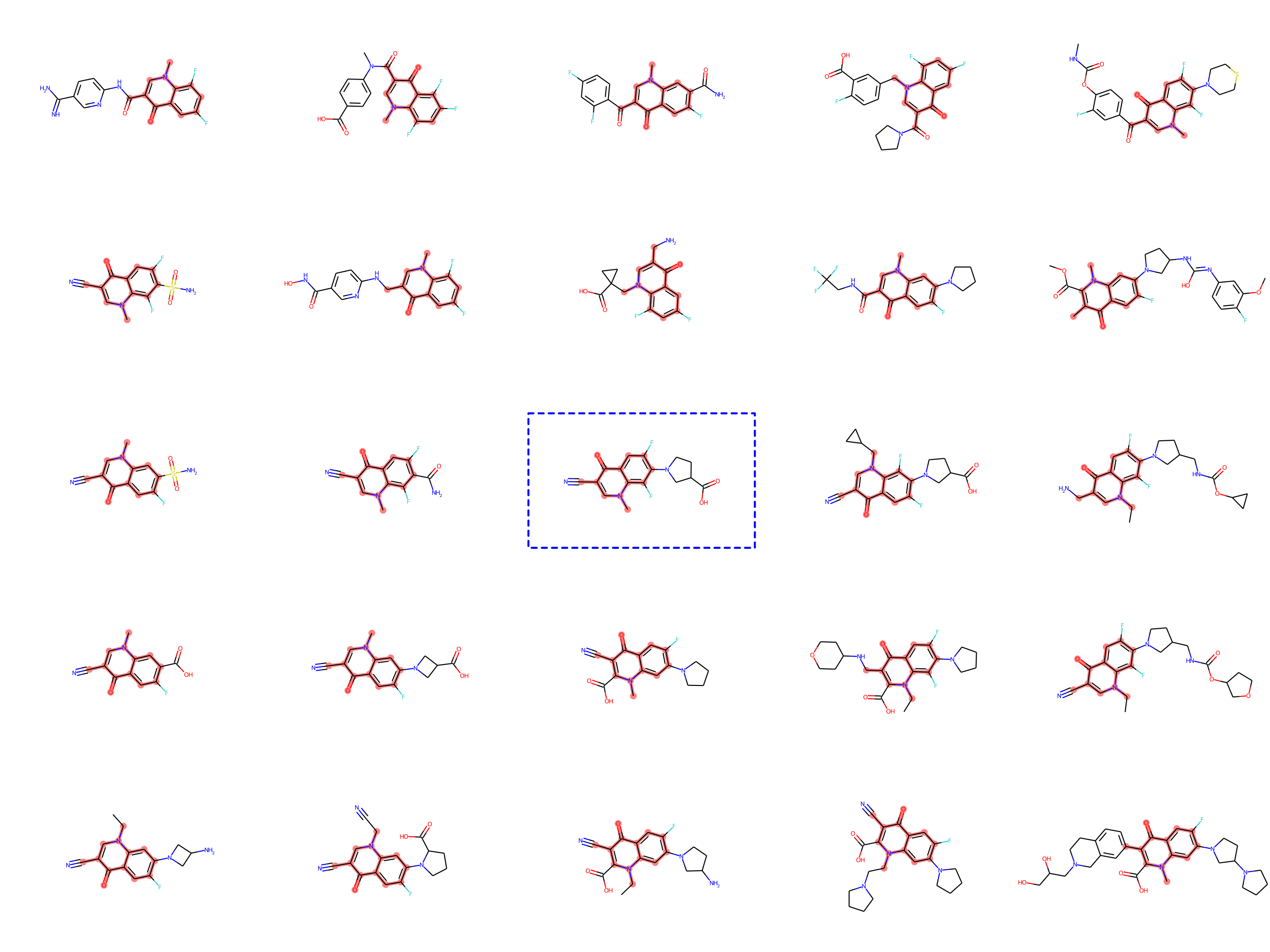
Optimization with MoLeR
Even after training our model as discussed above, MoLeR has no notion of “optimization” of molecules. However, like related approaches (opens in new tab), we can perform optimization in the space of latent codes using an off-the-shelf black-box optimization algorithm. This was not possible with CGVAE, which used a much more complicated encoding of graphs. In our work, we opted for using Molecular Swarm Optimization (MSO) (opens in new tab), which shows state-of-the-art results for latent space optimization in other models, and indeed we found it to work very well for MoLeR. In particular, we evaluated optimization with MSO and MoLeR on new benchmark tasks that are similar to realistic drug discovery projects using large scaffolds and found this combination to outperform existing models.
Spotlight: On-demand video

AI Explainer: Foundation models and the next era of AI
Explore how the transformer architecture, larger models and more data, and in-context learning have helped advance AI from perception to creation.
Outlook
We continue to work with Novartis to focus machine learning research on problems relevant to the real-world drug discovery process. The early results are substantially better than those of competing methods, including our earlier CGVAE model. With time, we hope MoLeR-generated compounds will reach the final stages of drug-discovery projects, eventually contributing to new useful drugs that benefit humanity.



