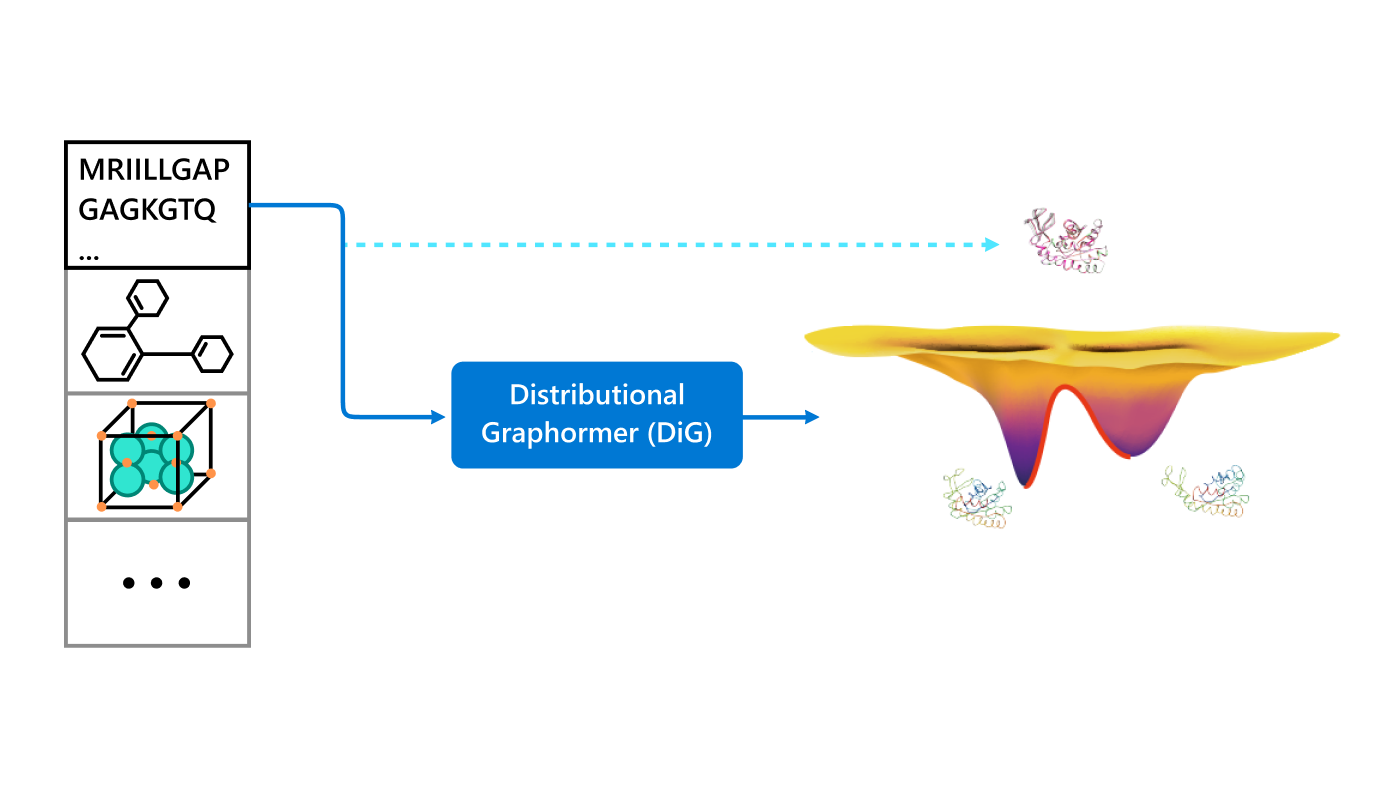
The essence of the biological world lies in the ever-changing nature of its molecules and their interactions. Understanding the dynamics and interactions of biomolecules is crucial for deciphering the mechanisms behind biological processes and for developing biomaterials and drugs. As Richard Feynman famously said, “Everything that living things do can be understood in terms of the jigglings and wigglings of atoms.” Yet capturing these real-life movements is nearly impossible through experiments.
In recent years, with the development of deep learning methods represented by AlphaFold and RoseTTAFold, predicting the static crystal protein structures has been achieved with experimental accuracy (as recognized by the 2024 Nobel Prize in Chemistry). However, accurately characterizing dynamics at an atomic resolution remains much more challenging, especially when the proteins play their roles and interact with other biomolecules or drug molecules.
As one approach, Molecular Dynamics (MD) simulation combines the laws of physics with numerical simulations to tackle the challenge of understanding biomolecular dynamics. This method has been widely used for decades to explore the relationship between the movements of molecules and their biological functions. In fact, the significance of MD simulations was underscored when the classic version of this technique was recognized with a Nobel Prize in 2013 (opens in new tab) (opens in new tab), highlighting its crucial role in advancing our understanding of complex biological systems. Similarly, the quantum mechanical approach—known as Density Functional Theory (DFT)—received its own Nobel Prize in 1998 (opens in new tab) (opens in new tab), marking a pivotal moment in computational chemistry.
In MD simulations, molecules are modeled at the atomic level by numerically solving equations of motions that account for the system’s time evolution, through which kinetic and thermodynamic properties can be computed. MD simulations are used to model the time-dependent motions of biomolecules. If you think of proteins like intricate gears in a clock, AI2BMD doesn’t just capture them in place—it watches them spin, revealing how their movements drive the complex processes that keep life running.
MD simulations can be roughly divided into two classes: classical MD and quantum mechanics. Classical MD employs simplified representations of the molecular systems, achieving fast simulation speed for long-time conformational changes but less accurate. In contrast, quantum mechanics models, such as Density Functional Theory, provide ground-up calculations, but are computationally prohibitive for large biomolecules.
Ab initio biomolecular dynamics simulation by AI
Microsoft Research has been working on the development of efficient methods aiming for ab initio accuracy simulations of biomolecules. This method, AI2BMD (AI-based ab initio biomolecular dynamics system), has published in the journal Nature (opens in new tab), representing the culmination of a four-year research endeavor.
AI2BMD efficiently simulates a wide range of proteins in all-atom resolution with more than 10,000 atoms at an approximate ab initio—or first-principles—accuracy. It thus strikes a previously inaccessible tradeoff for biomolecular simulations than standard simulation techniques – achieving higher accuracies than classical simulation, at a computational cost that is higher than classical simulation but orders of magnitude faster than what DFT could achieve. This development could unlock new capabilities in biomolecular modeling, especially for processes where high accuracy is needed, such as protein-drug interactions.
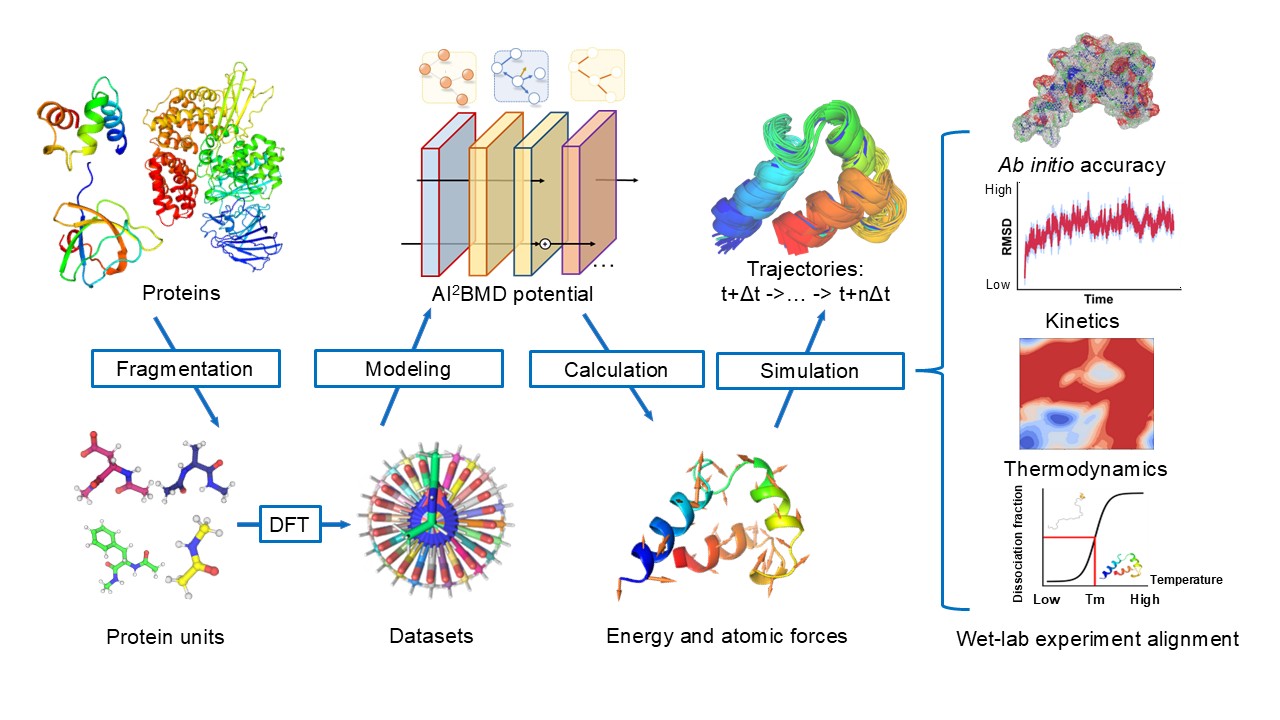
AI2BMD employs a novel-designed generalizable protein fragmentation approach that splits proteins into overlapping units, creating a dataset of 20 million snapshots—the largest ever at the DFT level. Based on our previously designed ViSNet (opens in new tab), a universal molecular geometry modeling foundation model published in Nature Communications (opens in new tab) and incorporated into PyTorch Geometry library (opens in new tab), we trained AI2BMD’s potential energy function using machine learning. Simulations are then performed by the highly efficient AI2BMD simulation system, where at each step, the AI2BMD potential based on ViSNet calculates the energy and atomic forces for the protein with ab initio accuracy. By comprehensive analysis from both kinetics and thermodynamics, AI2BMD exhibits much better alignments with wet-lab data, such as the folding free energy of proteins and different phenomenon than classic MD.
Microsoft research podcast

Collaborators: Silica in space with Richard Black and Dexter Greene
College freshman Dexter Greene and Microsoft research manager Richard Black discuss how technology that stores data in glass is supporting students as they expand earlier efforts to communicate what it means to be human to extraterrestrials.
Advancing biomolecular MD simulation
AI2BMD represents a significant advancement in the field of MD simulations from the following aspects:
(1) Ab initio accuracy: introduces a generalizable “machine learning force field,” a machine learned model of the interactions between atoms and molecules, for full-atom protein dynamics simulations with ab initio accuracy.
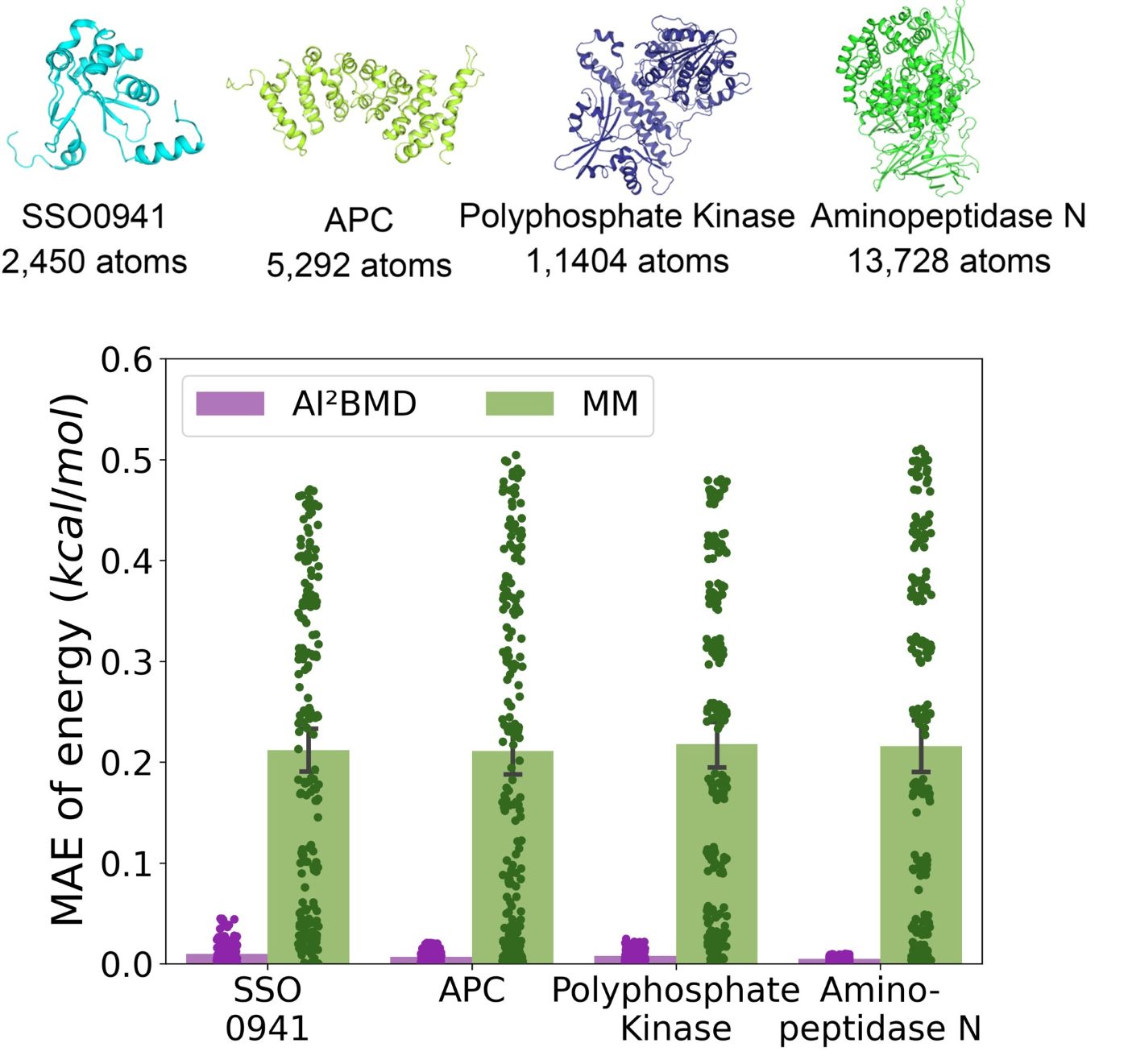
(2) Addressing generalization: It is the first to address the generalization challenge of a machine learned force field for simulating protein dynamics, demonstrating robust ab initio MD simulations for a variety of proteins.
(3) General compatibility: AI2BMD expands the Quantum Mechanics (QM) modeling from small, localized regions to entire proteins without requiring any prior knowledge on the protein. This eliminates the potential incompatibility between QM and MM calculations for proteins and accelerates QM region calculation by several orders of magnitude, bringing near ab initio calculation for full-atom proteins to reality. Consequently, AI2BMD paves the road for numerous downstream applications and allows for a fresh perspective on characterizing complex biomolecular dynamics.
(4) Speed advantage: AI2BMD is several orders of magnitude faster than DFT and other quantum mechanics. It supports ab initio calculations for proteins with more than 10 thousand atoms, making it one of the fastest AI-driven MD simulation programs among multidisciplinary fields.
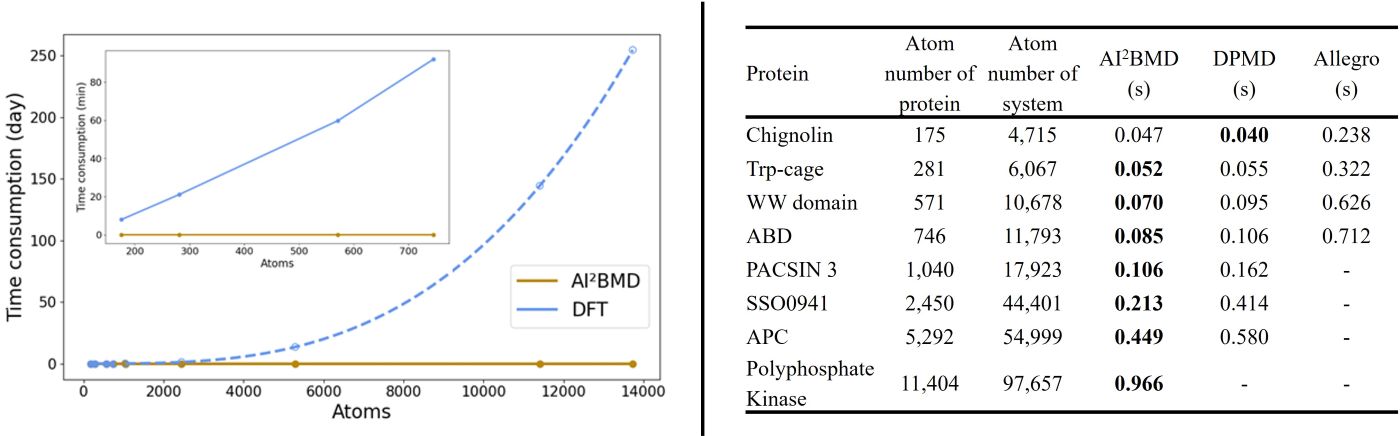
(5) Diverse conformational space exploration: For the protein folding and unfolding simulated by AI2BMD and MM, AI2BMD explores more possible conformational space that MM cannot detect. Therefore, AI2BMD opens more opportunities to study flexible protein motions during the drug-target binding process, enzyme catalysis, allosteric regulations, intrinsic disorder proteins and so on, better aligning with the wet-lab experiments and providing more comprehensive explanations and guidance to biomechanism detection and drug discovery.
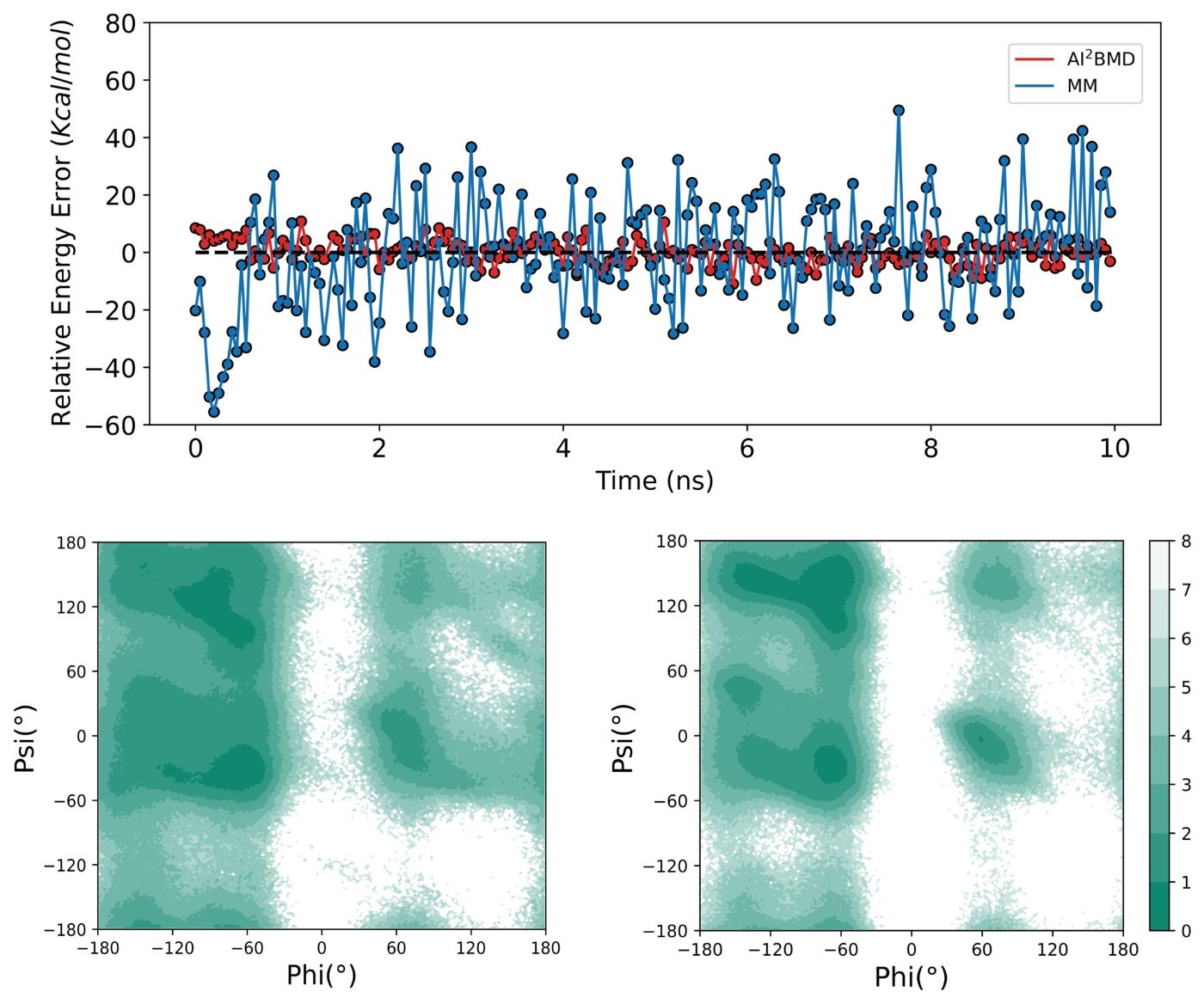
(6) Experimental agreement: AI2BMD outperforms the QM/MM hybrid approach and demonstrates high consistency with wet-lab experiments on different biological application scenarios, including J-coupling, enthalpy, heat capacity, folding free energy, melting temperature, and pKa calculations.
Looking ahead
Achieving ab initio accuracy in biomolecular simulations is challenging but holds great potential for understanding the mystery of biological systems and designing new biomaterials and drugs. This breakthrough is a testament to the vision of AI for Science—an initiative to channel the capabilities of artificial intelligence to revolutionize scientific inquiry. The proposed framework aims to address limitations regarding accuracy, robustness, and generalization in the application of machine learning force fields. AI2BMD provides generalizability, adaptability, and versatility in simulating various protein systems by considering the fundamental structure of proteins, namely stretches of amino acids. This approach enhances energy and force calculations as well as the estimation of kinetic and thermodynamic properties.
One key application of AI2BMD is its ability to perform highly accurate virtual screening for drug discovery. In 2023, at the inaugural Global AI Drug Development competition (opens in new tab), AI2BMD made a breakthrough by predicting a chemical compound that binds to the main protease of SARS-CoV-2. Its precise predictions surpassed those of all other competitors, securing first place and showcasing its immense potential to accelerate real-world drug discovery efforts.
Since 2022, Microsoft Research also partnered with the Global Health Drug Discovery Institute (GHDDI), a nonprofit research institute founded and supported by the Gates Foundation, to apply AI technology to design drugs that treat diseases that unproportionally affect low- and middle- income countries (LMIC), such as tuberculosis and malaria. Now, we have been closely collaborating with GHDDI to leverage AI2BMD and other AI capabilities to accelerate the drug discovery process.
AI2BMD can help advance solutions to scientific problems and enable new biomedical research in drug discovery, protein design, and enzyme engineering.