

作者:科学智能中心
编者按:随着科技的不断发展,新材料已成为增强产业竞争力的关键因素。然而,新材料的物理和化学特性复杂多变,准确预测其属性,特别是实际合成和使用条件下的属性,是物质科学领域中长期存在的挑战,也是材料工业数字化转型的核心挑战之一。
为了破解这一难题,微软研究院科学智能中心(Microsoft Research AI for Science)开发了深度学习模型 MatterSim,能够在广泛的元素、温度和压力范围内实现准确高效的材料模拟与性质预测,为材料设计的数字化转型提供了强有力的支持。

新材料探索对纳米电子学、能量储存和医疗健康等多个领域的技术进步至关重要。材料设计中的一个核心难点是如何在不进行实际合成和测试的情况下预测材料属性。由于新材料可能涉及元素周期表中118种元素的任意组合,且其合成和工作温度、压力范围极广,这些因素极大地影响了材料内部原子的相互作用,使得准确预测材料属性和行为模拟变得极为困难。
为此,微软研究院科学智能中心(Microsoft Research AI for Science)推出了 MatterSim 模型。该模型将深度学习技术和大规模第一性原理计算相结合来学习原子之间的相互作用,学习的材料空间从绝对零度到5000开尔文,从标准大气压到一千万倍大气压,能够高效处理多种材料的模拟,包括但不限于金属、氧化物、硫化物、卤化物及其不同状态(如晶体、非晶固体和液体)。此外,MatterSim 还提供定制选项,可以通过整合下游场景的数据来执行更为复杂的预测任务。
MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures
https://arxiv.org/abs/2405.04967 (opens in new tab)
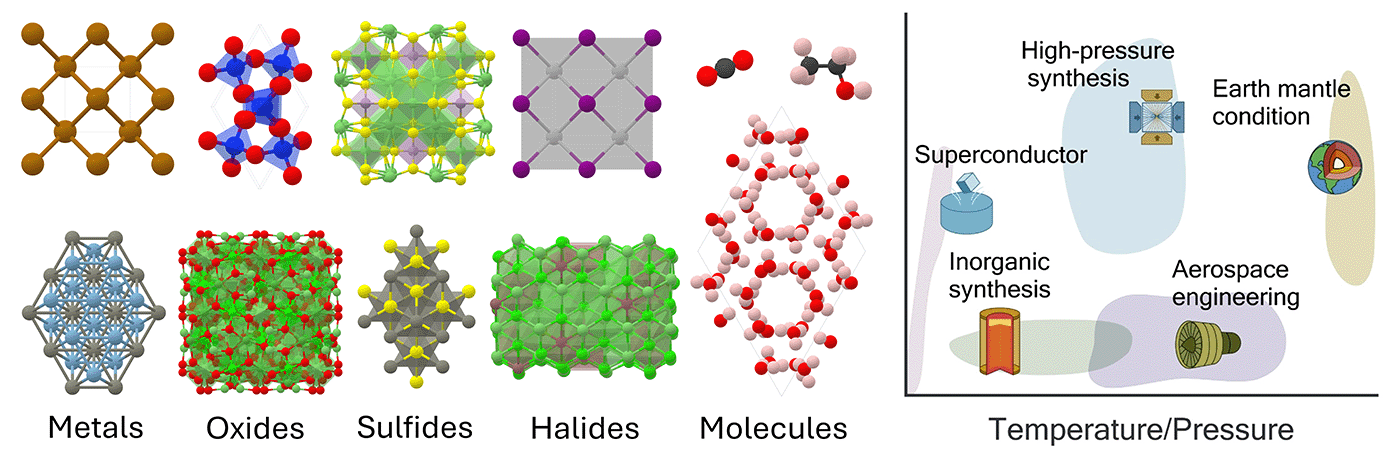
全周期表、跨温度、跨压力的真实条件下的材料模拟
MatterSim 的训练过程使用了大规模的合成数据。为了获得这些训练数据,研究员们结合了主动学习、分子动力学模拟和生成模型等技术,构建了高效的数据生成方案。这种数据生成策略确保了模型对材料空间的广泛覆盖,使其能够以与第一性原理预测相当的准确度,预测材料在原子层面的能量、力和应力。
与当前的 SOTA 模型相比,MatterSim 在有限的温度和压力下对材料属性预测的准确度提高了10倍。研究表明,MatterSim 能够准确模拟包括热力学、力学和运输特性在内的广泛材料属性。值得一提的是,MatterSim 可以从头构建材料相图,为新材料的可合成性与合成条件提供指导。

定制化精调:用更少的训练数据达到实验精度的模拟
尽管基于大量的合成数据集进行训练,MatterSim 可以通过整合额外的数据来满足特定的设计要求。MatterSim 能够利用主动学习和微调,定制化完成特定场合下高度复杂的材料模拟和设计任务。
以物质计算领域的经典任务——液态水性质的模拟为例,这个任务看似简单,实际上却需要大量的计算。通过 MatterSim 的定制化功能对该任务进行优化,MatterSim 只需要3%的原始数据,就能达到预期的实验精度模拟。相比之下,用传统方法训练的专有机器学习模型,则需要提供30倍的资源,而第一性原理的方法则需要增加指数级的资源。
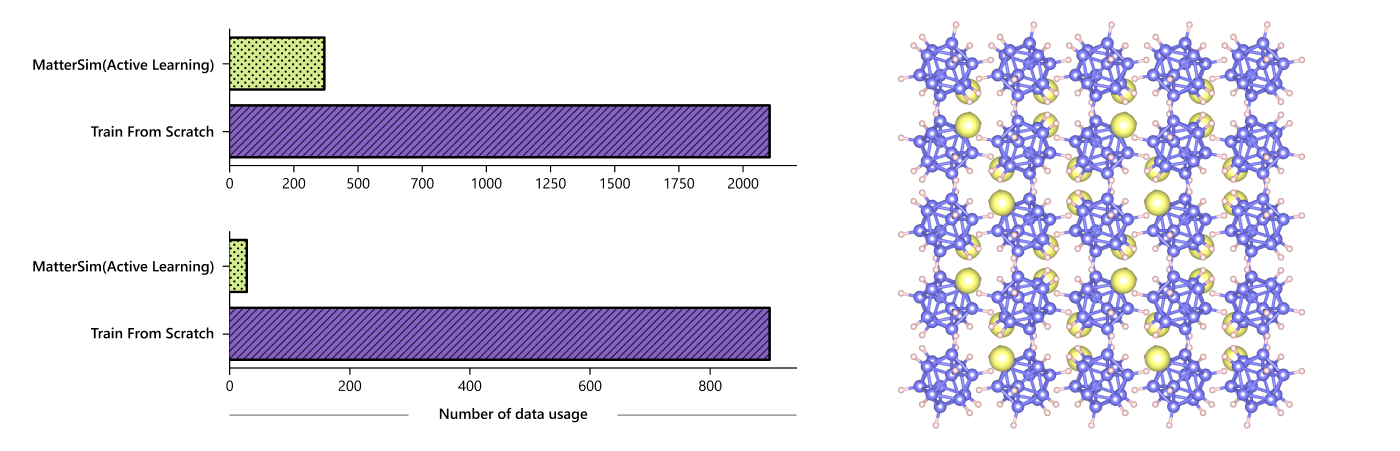
弥合材料微观模型与宏观属性间的鸿沟
从原子结构预测材料宏观属性是一项复杂的任务,对于目前基于统计力学的方法,如分子动力学,太过复杂。MatterSim 通过深度学习技术直接映射这些关系来解决这个问题。研究员们为 MatterSim 专门设计了可以微调的适配器模块,可以直接从结构数据预测材料属性,避免了复杂模拟的需求。在著名的材料属性预测基准测试集 MatBench 上的基准测试中,MatterSim 的精确度有了显著提升,并优于所有针对特定属性的专有模型,展现了其直接从领域特定数据预测材料属性的强大能力。
开启人工智能辅助材料设计新篇章
MatterSim 为鲁棒且高效的材料模拟和性质预测提供了新的可能性,其与 Distributional Graphormer 等生成式人工智能技术和强化学习技术的结合,将有望彻底改变材料科学研究的理念与模式。以 MatterSim、MatterGen 为代表的材料科学与人工智能模型,也将使定制化材料的开发变得更加高效,应用范围更广,从半导体技术到生物医药工程等诸多领域都将从中受益。
未来,研究员们将继续推进 MatterSim 的实验验证,希望相关研究可以在面向可持续发展的催化剂设计、能源存储、纳米技术等领域发挥作用,造福全球社会。
相关链接: