

作者:科学智能中心
编者按:为了使电子结构方法突破当前广泛应用的密度泛函理论(KSDFT)所能求解的分子体系规模,微软研究院科学智能中心的研究员们基于人工智能技术和无轨道密度泛函理论(OFDFT)开发了一种新的电子结构计算框架 M-OFDFT。这一框架不仅保持了与 KSDFT 相当的计算精度,而且在计算效率上实现了显著提升,并展现了优异的外推性能,为分子科学研究中诸多计算方法的基础——电子结构方法开辟了新的思路。相关研究成果已在国际知名学术期刊《自然-计算科学》(Nature Computational Science)上发表。
近几十年来,理论与计算化学领域取得的一大成就是能够通过计算手段得到分子体系的物理化学性质。这为药物发现和材料设计等诸多工业界问题带来了全新的研究手段,有望缩短开发流程并降低开发成本。这些计算方法的基础步骤是使用电子结构方法求解给定分子体系的电子状态,进而得到该体系的各种性质。
然而,各种电子结构方法的求解精度和计算效率往往无法兼得。当前,取得相对合理的“精度-效率”权衡而被广泛应用的方法是 Kohn-Sham 形式的密度泛函理论(Kohn-Sham density functional theory, KSDFT)。但 KSDFT 具有较高的计算复杂度,不能很好地满足日益增长的求解大规模分子体系的需求。为此,微软研究院科学智能中心的研究员们提出了一种基于深度学习和无轨道密度泛函理论(OFDFT)的电子结构计算框架 M-OFDFT,其不仅显著超越了 KSDFT 的计算效率,还能保有其求解精度。这一成果展示了人工智能在提升电子结构计算中“精度-效率”权衡方面的卓越能力,并将助力加速相关业界问题的研究与开发。M-OFDFT 的相关研究成果已在国际知名学术期刊《自然-计算科学》(Nature Computational Science)上发表。

Overcoming the Barrier of Orbital-Free Density Functional Theory for Molecular Systems Using Deep Learning
《自然-计算科学》文章链接:https://www.nature.com/articles/s43588-024-00605-8 (opens in new tab)
SharedIt 链接:https://rdcu.be/dANtS (opens in new tab)
论文链接:https://arxiv.org/abs/2309.16578 (opens in new tab)
人工智能给电子结构方法带来新机会
电子结构方法是求解分子体系各种物理化学性质的基础工具。由于多电子体系本身具有一定的求解难度,所以高精度电子结构方法因其较高的计算代价很难应用到工业界所关注的分子体系中,而可计算较大分子的方法则会因引入一些近似而损失精度。目前 KSDFT 因其相对合适的精度与效率权衡得到了广泛应用。
不过,近期人工智能技术的喜人进展也为其他电子结构计算框架带来了新的机会。为了使电子结构方法突破 KSDFT 所能求解的分子体系规模,微软研究院的研究员们利用人工智能技术,开发了 M-OFDFT,该方法比 KSDFT 效率更高,同时又能保有其精度。基于 OFDFT 的开发,让 M-OFDFT 成为了一种比 KSDFT 理论复杂度更低的电子结构计算框架,因为它只需优化电子密度函数 ρ(r) 这一个函数来求解电子状态即可,KSDFT 则需要优化与电子数相同的多个函数。
不过,OFDFT 面临着一个巨大的挑战——需要电子动能关于密度函数的泛函 T_S [ρ],但它的形式未知,并且难以构造适用于分子体系的高精度近似。
针对这一难题,M-OFDFT 使用一个深度学习模型 T_(S,θ) 来近似动能泛函。借助深度学习模型的强大拟合能力,M-OFDFT 可实现比基于近似物理模型设计的经典动能泛函更高的准确度。对于一个待求解的分子体系结构,M-OFDFT 会使用动能泛函模型 T_(S,θ) 以及其他可直接计算的能量项构造出一个电子密度的优化目标,然后通过优化过程求解最优(基态)电子密度(图1),进而可计算能量、力、电荷分布等分子属性。

M-OFDFT实现兼具精度与效率的电子结构方法
研究员们对 M-OFDFT 进行了一系列的实验验证。首先考察的是 M-OFDFT 在常见小分子体系上的求解精度。结果显示,M-OFDFT 在乙醇分子构象以及 QM9 数据集的分子上可以达到与KSDFT相当的精度(能量达到化学精度1 kcal/mol)。相较于经典 OFDFT 方法,精度提高了两个数量级(图2-a)。M-OFDFT 解得的电子密度也与 KSDFT 的结果重合(图2-b),特别是得到了电子壳层结构,而经典 OFDFT 的结果则有明显偏差。由 M-OFDFT 解得的乙醇构象空间上的势能面(每个点都是通过密度优化得到的,并不是直接预测)也与 KSDFT 的结果一致(图2-c)。

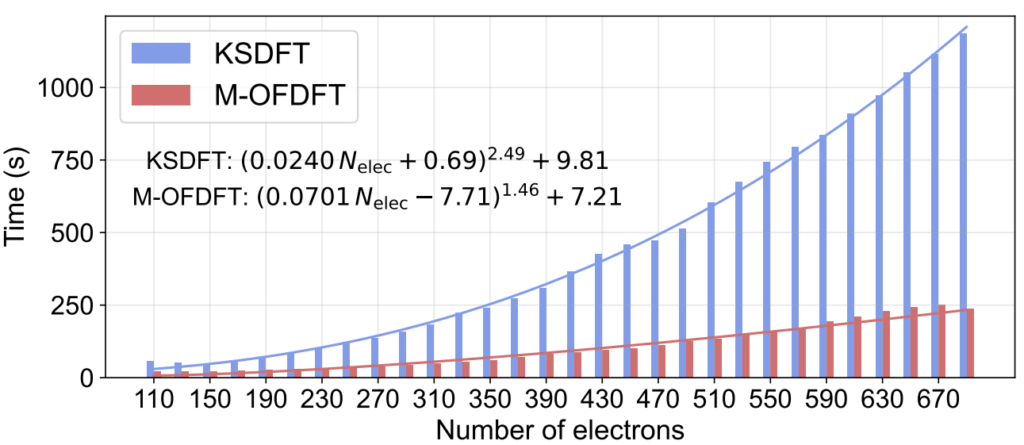
之后,研究员们又验证了 M-OFDFT 不仅保有 KSDFT 级别的精度,其更低的理论计算复杂度还使其在效率上也超越了 KSDFT。在实际计算中 M-OFDFT 取得了 O(N^1.46) 的复杂度(图3),比 KSDFT 的实际复杂度 O(N^2.49) 低了一阶,且其所需绝对时间也明显少于 KSDFT。在两个更大的蛋白质体系上(包含2676和2750个电子),M-OFDFT 实现了25.6倍和27.4倍的加速。
M-OFDFT具有更强的泛化能力
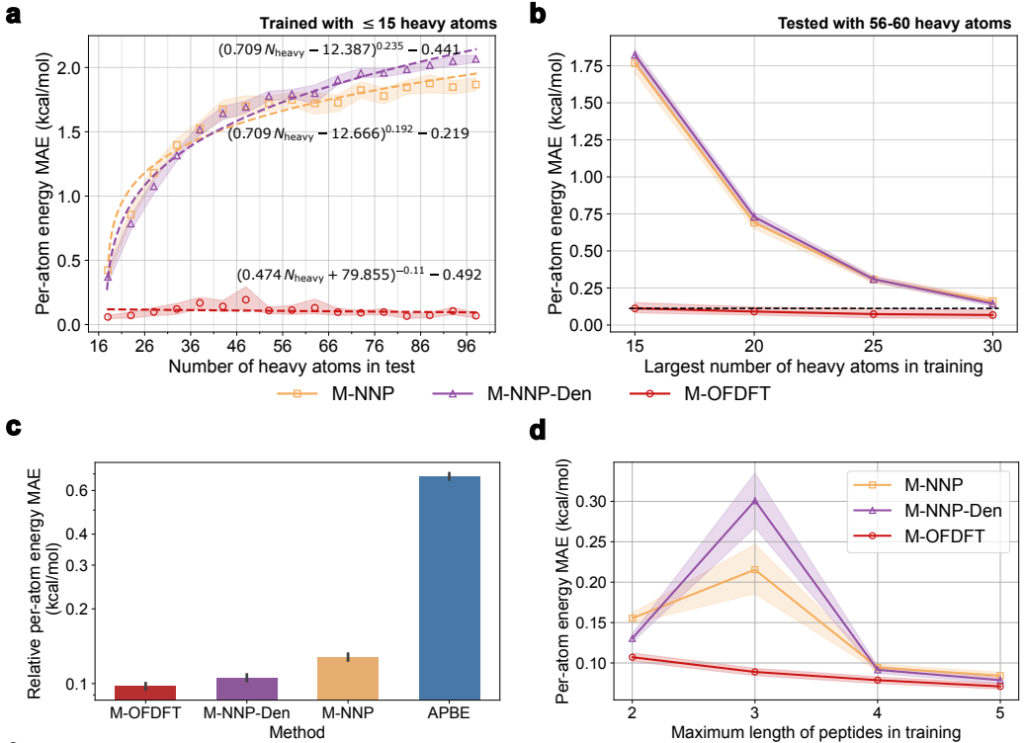
深度学习模型在科学任务中的应用面临一大挑战是,在具有与训练数据不同特点的数据上的泛化问题。但采用了 OFDFT 框架后,动能泛函模型遇到的泛化问题就会减轻,从而使 M-OFDFT 可以在比训练集分子规模更大的体系上展现出良好的外推能力。
实验结果表明,M-OFDFT 的能量预测误差显著低于基于深度学习的端到端能量预测模型(图4-a)。此外,研究员们还利用在多肽片段上训练的 M-OFDFT 模型求解完整蛋白结构,并取得了超越端到端模型和经典 OFDFT 的泛化性能(图4-c)。不仅如此,相较端到端模型,M-OFDFT 还可以用更少的大分子体系训练数据取得更好的泛化表现(图4-b与图4-d)。
M-OFDFT的工作原理
“神龙见首又见尾”:高效捕获非局域效应的动能泛函模型
动能密度泛函具有明显的非局域效应,而用经典的基于格点(grid)的方式表征电子密度则会带来高昂的非局域计算代价。为此,M-OFDFT 将电子密度在一组原子基组函数上展开,并使用展开系数 p 作为电子密度表征。由于基函数叠加的形状与电子分布接近,所以其数量可远小于格点数,使得非局域计算代价大大降低,并有助于刻画电子密度中的壳层结构。
M-OFDFT 将每个原子上的电子密度系数 p 和类型 Z 与坐标 x 作为节点特征,并基于 Graphormer 模型[1]预测电子动能 T_(S,θ)(图5),其自注意力机制显式刻画了荷载在每两个原子上的电子密度特征之间的相互作用,从而可捕捉非局域性质。此外,为了保证动能的旋转不变性,M-OFDFT 使用了以各个原子为中心、基于其相邻原子的局部坐标系,将电子密度系数转换为旋转不变的特征。

“横看成岭侧成峰,远近高低各不同”:高效学习电子能量曲面的训练策略
与传统机器学习任务不同,动能泛函模型是被当作其输入变量的优化目标使用的,而非用于在一些单点上做预测,这对模型的学习提出了更高的要求:模型必须捕捉到每个分子结构上电子能量曲面的轮廓。
为此,研究员们深入分析了用来生成数据的电子结构方法,发现它其实可以为每个分子结构生成多个数据点,而且还能提供梯度标注,从而让模型可以拥有更丰富的曲面轮廓特征。然而梯度的巨大范围也使神经网络难以优化。对此,研究员们还提出了一系列增强模块,让模型能够更容易地表达巨大的梯度。
开启未来电子结构方法的新篇章
M-OFDFT 成功突破了无轨道密度泛函框架在分子体系中的瓶颈,将其求解精度提升到了常用的 KSDFT 的水平,同时保有了其更低的计算代价,推进了电子结构方法在“精度-效率”方面的权衡,为分子科学研究提供了一种更有潜力的研究工具。
尽管 M-OFDFT 已经在某些分子体系上展现了出色的泛化性能,但在更大的分子体系上实现长时间且稳定的高精度模拟仍是一个巨大的挑战。微软研究院期待 M-OFDFT 可以沿着这一方向激发更多研究与创新,并在未来和其他方法一起为电子结构计算带来更多突破性的成果和影响。
相关文章
[1] Do Transformers really perform badly for graph representation? Advances in Neural Information Processing Systems 34 (NeurIPS 2021)