

作者:科学智能中心
编者按:分子动力学模拟在新药开发、材料设计等领域发挥着重要作用。近年来机器学习技术的不断发展,使得其对分子间相互作用的刻画也更加精确,但却面临着随分子体系扩大,计算效率降低和长程信息丢失的难题。在此背景下,微软研究院科学智能中心同耶鲁大学、西安交通大学提出了一种名为 LSR-MP 的新型分子动力学机器学习框架。该框架结合了物理洞见和几何深度学习,通过在原子/分子片段上分别建模短程和长程效应,为大规模分子体系的高精度、高效模拟开辟了新的途径。
分子动力学模拟是研究分子体系的“显微镜”,在新药开发、材料设计等领域发挥着重要作用。但传统的模拟方法面临计算成本高、难以处理大分子等瓶颈。近年来,人工智能技术的发展为突破这些瓶颈带来了新的希望。
近日,微软研究院科学智能中心(Microsoft Research AI for Science)携手耶鲁大学、西安交通大学提出了一种名为 LSR-MP 的新型分子动力学机器学习框架。该框架巧妙融合了量子化学领域的分割思想与几何深度学习技术,在精准捕捉分子间相互作用的同时,大幅提升了计算效率。相关论文已被国际人工智能顶级学术会议之一的 ICLR 2024 接收。

论文链接:https://openreview.net/pdf?id=rvDQtdMnOl (opens in new tab)
探索多尺度相互作用建模的可扩展性
分子动力学模拟是通过求解物理方程,预测分子体系的时间演化过程,来获得其性质与功能的信息。传统的从头算方法,如密度泛函理论(DFT),虽然精度较高,但计算成本随分子尺寸呈O(N3)到O(N4)之间的多项式级增长,难以应用于蛋白质等大分子体系。
近年来,研究人员尝试利用机器学习来“学习”分子间的相互作用,从而加速模拟。这其中,图神经网络(GNNs)凭借强大的图结构数据处理能力崭露头角。特别是等变图神经网络(EGNNs)通过编码分子体系的物理对称性,在小分子数据集上展现出了优越的性能。然而,当把它们用于数百个原子的生物大分子时,研究人员发现长程相互作用(如静电作用、范德华力)的信息容易丢失,影响模型性能,而且简单增大感受野或加深网络,会导致计算效率降低、过拟合、过度挤压等问题发生。
如何在建模分子间多尺度相互作用的同时,保持模型的可扩展性,是一个亟待解决的难题。
在过去几年,基于分子分割的量子化学方法和机器学习方法为大型分子体系的模拟带来了新的曙光。基于分割的方法,如分治法(Divide-and-Conquer,DC)和基于共轭帽的分子分割法(Molecular Fractionation with Conjugated Caps,MFCC)可以将体系切割为易处理的小片段,在一定程度上降低了计算复杂度。然而,这类方法在描述片段间的多体相互作用时还不够精确,限制了它们的进一步应用。
因此,一种既能像基于分割的方法那样处理大体系,又能像等变神经网络那样精准学习原子间复杂相互作用的新的机器学习框架亟需开发。LSR-MP框架由此诞生。
LSR-MP:融合物理洞见的分子动力学学习范式
LSR-MP 框架结合了物理洞见和几何深度学习,可以看作是对现有 EGNNs 的扩展。如图1所示,该框架的核心在于引入了分子片段的概念,并在原子/分子片段两个层次上分别建模短程和长程效应。
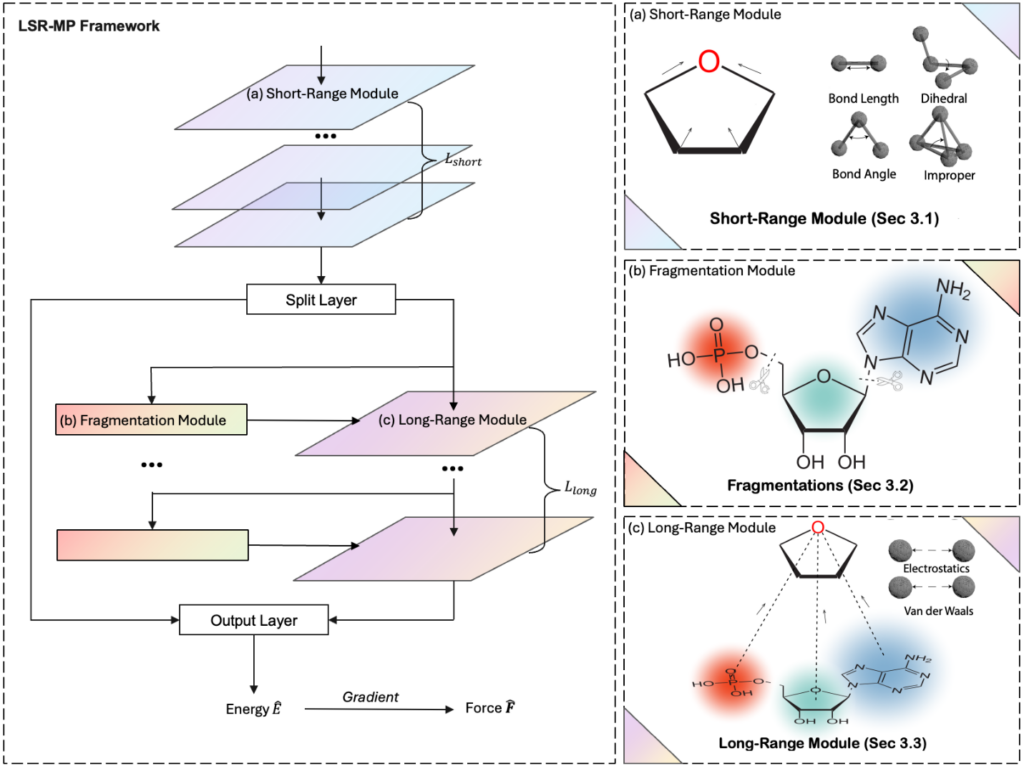
具体而言,LSR-MP 框架包含三个关键模块:短程模块、分割模块和长程模块。短程模块会采用 EGNNs,在原子的局部邻域内传递信息,捕捉化学键、键角等几何信息。这一过程可以看作在由短程截断半径定义的子图上进行消息传递。分割模块则会使用 BRICS (Breaking of Retrosynthetically Interesting Chemical Substructures)算法,基于化学启发式规则,将分子非重叠地划分为多个片段。每个片段的表示会通过求和其包含的原子表示而得到。本质上,分割模块是在原子和片段的粒度间建立映射。而长程模块主要负责捕捉片段间的相互作用。研究员们构建了一个原子-分子片段的二部图,其中原子和分子片段之间的边由一个较大的长程截断半径定义。在此二部图上,研究员们还设计了方向性注意力机制,用于长程信息传递。
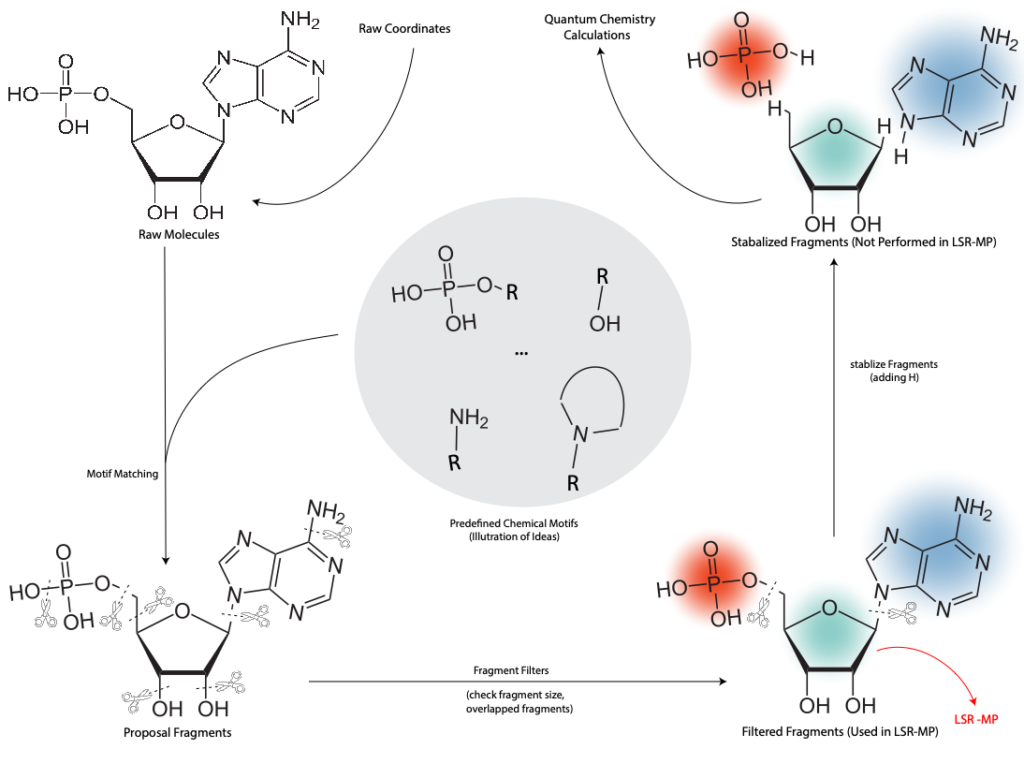
- BRICS 是一种基于规则的化学分割算法,如图2所示,常用于将大型分子划分为合适的片段,以便进行量子化学计算或机器学习建模。其基本原理如下:
- 预定义化学键模式:BRICS 预定义了16种化学键模式,这些模式是由有机化学专家根据经验总结的,代表了分子中相对容易断裂的键。例如,碳-杂原子键,苄位 C-C 键等。
- 断键:对于输入的分子,BRICS 先识别其中符合预定义模式的化学键,将分子按照这些键断开,来得到一系列片段。
- 片段修饰:断键后的片段通常带有悬挂键,BRICS 会在断键处加上合适的原子(通常是氢原子),以使得片段在化学上稳定。
- 片段过滤:BRICS 还会对生成的片段进行后处理,包括去除过小的片段(如单个原子)、去除重复片段等,以保证片段集合的合理性。
与其他基于图的分割算法相比,BRICS 具有显著优势。它不仅能够利用化学家经验中预定义的化学键模式,生成合理且符合化学直觉的片段,而且能够在分割过程中最大程度地保留分子的化学特征,使得生成的片段在后续建模中更具代表性。此外,BRICS 作为一种基于规则的算法,其计算效率较高,特别适合处理大规模数据集。
BRICS 算法在片段生成领域被广泛使用,并在多个分子模拟任务中取得了不错的效果。在 LSR-MP 框架中,BRICS 扮演着重要的角色,负责在原子-片段两个粒度之间建立联系,为长短程信息传递提供基础。
通过短程模块处理局部相互作用,并用长程模块补足长程相互作用,LSR-MP 框架在处理大型分子体系时,平衡了计算效率和物理准确性。同时,分割模块充当了原子/分子片段粒度之间的桥梁,使该框架能够灵活处理不同的尺度体系。
根据研究,LSR-MP 框架在数学上还具有等变性,这意味着它能很好地适应分子体系的平移、旋转等对称性,而这是精准分子模拟的关键。此外,通过引入基于片段的长程信息传递,LSR-MP 框架能够缓解图神经网络中的过度挤压问题,提高深层网络的学习能力。
高效能预测,跨模型性能提升
研究员们在 MD22 和 Chignolin 两个广泛使用的基准数据集上,将基于 LSR-MP 的 ViSNet-LSRM 模型与多个 SOTA 模型进行了系统比较,其实验结果均提升显著。
ViSNet-LSRM 在大多数分子的能量和力预测任务上,都取得了最优表现。以114个原子组成的 AT-AT-CG-CG 分子为例,ViSNet-LSRM 将力预测误差降至0.11 kcal/mol·Å,较优于 Equiformer 等模型15-40%。在166个原子的 Chignolin 蛋白上,ViSNet-LSRM 的能量预测误差比主流模型低了30%以上。
更重要的是,ViSNet-LSRM 在模型效率上同样表现优秀。它的参数量仅为 Equiformer 的56%,达到了与之相当甚至更优的预测精度。推理速度上,ViSNet-LSRM 也比主流模型快了8~43倍。
此外,研究员们考察了 LSR-MP 框架的可拓展性(图3)。实验表明,当将 LSR-MP 集成到 PaiNN、Equiformer 等不同的 GNN 模型后,它们的预测误差平均降低了30%以上。这证实了 LSR-MP 引入的物理信息具有一定的“普世价值”,能适用于不同的模型。

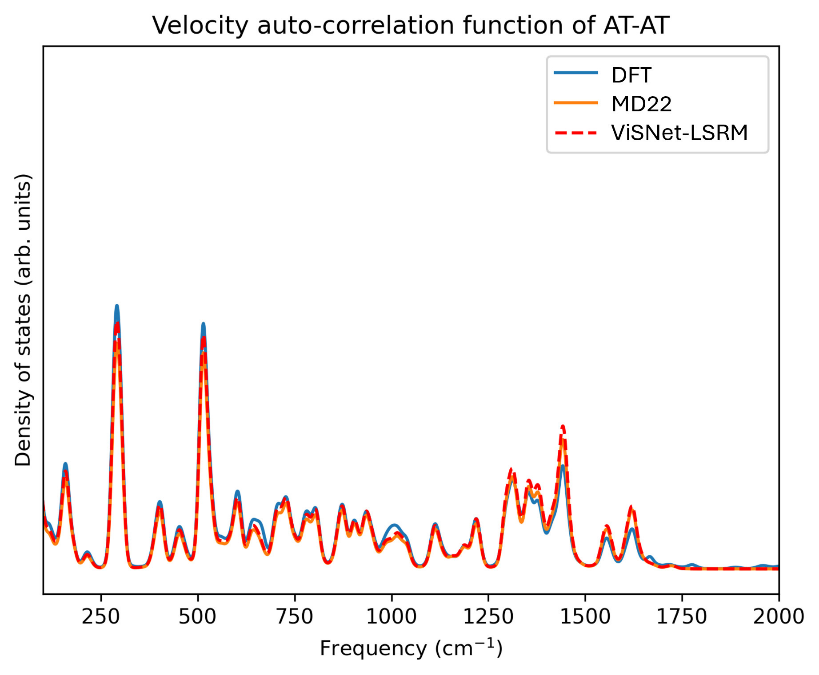
最后,研究员们对一个核苷酸分子体系(AT-AT)进行了 20ps 的分子动力学模拟(单步步长为1fs),使用 ViSNet-LSRM 作为力场,并在恒能量集合(NVE)条件下进行。通过分析 AT-AT 分子的振动光谱(如图4所示)可以看到,尽管与 DFT 结果相比存在微小的峰强度差异,但 MD22 数据集中的轨迹与 ViSNet-LSRM 模拟的轨迹展现出了相似的振动光谱。这一发现表明,LSR-MP 的模拟能够准确再现分子在较长时间尺度上的实际振动模式。
展望未来:拓展更广泛的物理化学过程
LSR-MP 框架为大规模分子体系的高精度、高效模拟开辟了新的途径。通过从量化计算中提炼物理规律(如化学局域性),并将其融入到 GNNs 的归纳偏置(如旋转平移等变性)中,LSR-MP 在经验性和物理可解释性间实现了平衡,为构建符合科学规律的 AI 模型提供了思路。
论文通讯作者、微软亚洲研究院科学智能中心高级研究员黄麟表示:“我们期待 LSR-MP 所倡导的‘知识引导的 AI 范式’可以在更多科学和工程领域开花结果,催生出更多像 LSR-MP 这样既有创新性,又兼顾物理可解释性的 AI 模型。”
未来,研究员们将进一步拓展 LSR-MP,用于模拟化学反应、材料缺陷等更广泛的物理化学过程。方法上,也将探索可学习的自适应分割策略,赋予模型“自我优化”的能力。站在分子模拟与 AI 技术交叉的前沿,LSR-MP 框架有望为加速药物研发、新材料设计带来新的推动力。